
The U.S. Food and Drug Administration says it wants to improve transparency around medical device safety, yet the agency is still allowing device companies to file secretive reports of deaths involving novel, permanent implants in the heart.
Earlier this year, the FDA ended a little-noticed but long-running system known as "alternative summary reporting" (ASR), which previously had let device companies avoid publicly filing full narrative reports about specific devices that the agency felt were well-understood.
The ASR program was ended after then-FDA Commissioner Dr. Scott Gottlieb tweeted last March that it was "imperative that all safety information be available to the public." (Gottlieb left the agency the following month.)
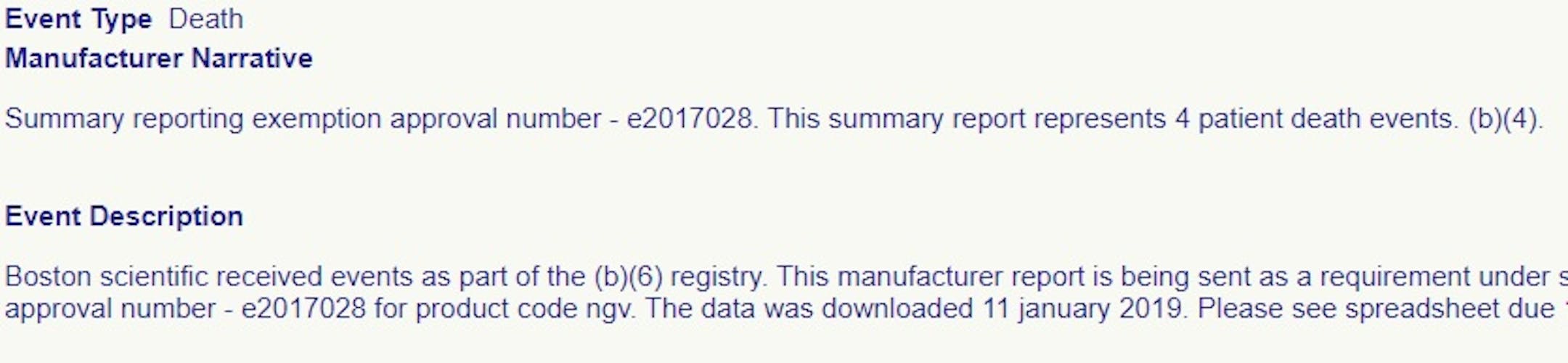
Yet last week, the FDA declined to release any information about four deaths summarized in a report about Boston Scientific's Watchman left-atrial appendage closure device -- a novel, permanent heart implant used to cut stroke risk in patients with atrial fibrillation.
And in October, a research letter published in JAMA Internal Medicine said the FDA was still accepting secretive "summary" reports of problems involving Edwards Lifesciences' Sapien 3 aortic heart valve and Abbott Laboratories' MitraClip system for mitral valve repair.
Though the FDA ended alternative summary reporting, it still allowed Boston Scientific to file an adverse event report that summarized four deaths in a few lines of text, and even kept secret the name of the underlying study that uncovered the deaths. FDA declined to release any further detail about the four deaths, including the "spreadsheet" mentioned in the public-facing adverse event report.
An FDA official said in an email that the secret Watchman reports were part of a different summary reporting system that the FDA continues to operate – even though FDA officials are aware that granting such exemptions will lead to further non-public reporting of medical device events.
Last year, the FDA said it was making changes in its device-surveillance division to "free up resources to better focus on addressing the highest risks, such as deaths and serious injuries, associated with medical devices."


